当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Modeling Insight into Battery Electrolyte Electrochemical Stability and Interfacial Structure
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2017-11-22 00:00:00 , DOI: 10.1021/acs.accounts.7b00486 Oleg Borodin 1 , Xiaoming Ren 1 , Jenel Vatamanu 1 , Arthur von Wald Cresce 1 , Jaroslaw Knap 2 , Kang Xu 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2017-11-22 00:00:00 , DOI: 10.1021/acs.accounts.7b00486 Oleg Borodin 1 , Xiaoming Ren 1 , Jenel Vatamanu 1 , Arthur von Wald Cresce 1 , Jaroslaw Knap 2 , Kang Xu 1
Affiliation
|
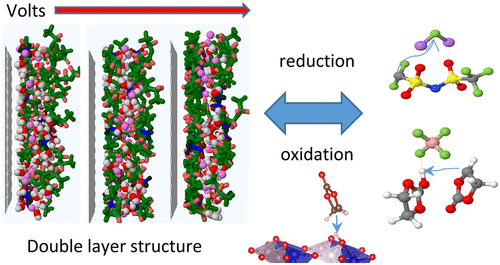
Electroactive interfaces distinguish electrochemistry from chemistry and enable electrochemical energy devices like batteries, fuel cells, and electric double layer capacitors. In batteries, electrolytes should be either thermodynamically stable at the electrode interfaces or kinetically stable by forming an electronically insulating but ionically conducting interphase. In addition to a traditional optimization of electrolytes by adding cosolvents and sacrificial additives to preferentially reduce or oxidize at the electrode surfaces, knowledge of the local electrolyte composition and structure within the double layer as a function of voltage constitutes the basis of manipulating an interphase and expanding the operating windows of electrochemical devices. In this work, we focus on how the molecular-scale insight into the solvent and ion partitioning in the electrolyte double layer as a function of applied potential could predict changes in electrolyte stability and its initial oxidation and reduction reactions. In molecular dynamics (MD) simulations, highly concentrated lithium aqueous and nonaqueous electrolytes were found to exclude the solvent molecules from directly interacting with the positive electrode surface, which provides an additional mechanism for extending the electrolyte oxidation stability in addition to the well-established simple elimination of “free” solvent at high salt concentrations. We demonstrate that depending on their chemical structures, the anions could be designed to preferentially adsorb or desorb from the positive electrode with increasing electrode potential. This provides additional leverage to dictate the order of anion oxidation and to effectively select a sacrificial anion for decomposition. The opposite electrosorption behaviors of bis(trifluoromethane)sulfonimide (TFSI) and trifluoromethanesulfonate (OTF) as predicted by MD simulation in highly concentrated aqueous electrolytes were confirmed by surface enhanced infrared spectroscopy.
中文翻译:

电池电解质电化学稳定性和界面结构的建模洞察
电活性界面将电化学与化学区分开来,并启用了诸如电池,燃料电池和双电层电容器之类的电化学能源设备。在电池中,电解质应在电极界面处保持热力学稳定,或通过形成电绝缘但离子导电的界面来保持动力学稳定。除了通过添加助溶剂和牺牲性添加剂来优先还原或氧化电极表面的传统电解质优化方法外,双层内局部电解质成分和结构作为电压函数的知识构成了操纵相间和膨胀的基础电化学设备的操作窗口。在这项工作中,我们专注于分子尺度对电解质双层中溶剂和离子分配的分子洞察力如何作为施加电势的函数,可以预测电解质稳定性及其初始氧化和还原反应的变化。在分子动力学(MD)模拟中,发现高浓度的锂水和非水电解质可将溶剂分子与正电极表面直接相互作用排除在外,这为扩展电解质的氧化稳定性提供了另外的机制,除了已建立的简单方法外消除高盐浓度下的“游离”溶剂。我们证明,根据其化学结构,可以将阴离子设计为随着电极电位的增加而优先从正电极吸附或解吸。这提供了额外的杠杆作用来决定阴离子的氧化顺序并有效地选择牺牲性阴离子进行分解。通过表面增强红外光谱证实了通过MD模拟预测的双(三氟甲烷)磺酰亚胺(TFSI)和三氟甲磺酸盐(OTF)的相反电吸附行为。
更新日期:2017-11-22
中文翻译:
电池电解质电化学稳定性和界面结构的建模洞察
电活性界面将电化学与化学区分开来,并启用了诸如电池,燃料电池和双电层电容器之类的电化学能源设备。在电池中,电解质应在电极界面处保持热力学稳定,或通过形成电绝缘但离子导电的界面来保持动力学稳定。除了通过添加助溶剂和牺牲性添加剂来优先还原或氧化电极表面的传统电解质优化方法外,双层内局部电解质成分和结构作为电压函数的知识构成了操纵相间和膨胀的基础电化学设备的操作窗口。在这项工作中,我们专注于分子尺度对电解质双层中溶剂和离子分配的分子洞察力如何作为施加电势的函数,可以预测电解质稳定性及其初始氧化和还原反应的变化。在分子动力学(MD)模拟中,发现高浓度的锂水和非水电解质可将溶剂分子与正电极表面直接相互作用排除在外,这为扩展电解质的氧化稳定性提供了另外的机制,除了已建立的简单方法外消除高盐浓度下的“游离”溶剂。我们证明,根据其化学结构,可以将阴离子设计为随着电极电位的增加而优先从正电极吸附或解吸。这提供了额外的杠杆作用来决定阴离子的氧化顺序并有效地选择牺牲性阴离子进行分解。通过表面增强红外光谱证实了通过MD模拟预测的双(三氟甲烷)磺酰亚胺(TFSI)和三氟甲磺酸盐(OTF)的相反电吸附行为。


























 京公网安备 11010802027423号
京公网安备 11010802027423号