PLoS Pathogens ( IF 6.7 ) Pub Date : 2017-11-20 , DOI: 10.1371/journal.ppat.1006702 Elsa Rousseau , Benoît Moury , Ludovic Mailleret , Rachid Senoussi , Alain Palloix , Vincent Simon , Sophie Valière , Frédéric Grognard , Frédéric Fabre
|
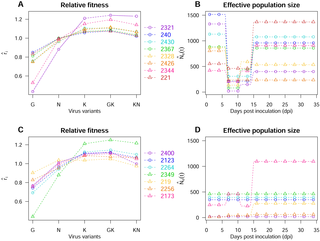
By combining high-throughput sequencing (HTS) with experimental evolution, we can observe the within-host dynamics of pathogen variants of biomedical or ecological interest. We studied the evolutionary dynamics of five variants of Potato virus Y (PVY) in 15 doubled-haploid lines of pepper. All plants were inoculated with the same mixture of virus variants and, variant frequencies were determined by HTS in eight plants of each pepper line, at each of six sampling dates. We developed a method for estimating the intensities of selection and genetic drift in a multi-allelic Wright-Fisher model, applicable whether these forces are strong or weak, and in the absence of neutral markers. This method requires variant frequency determination at several time points, in independent hosts. The parameters are the selection coefficients for each PVY variant and four effective population sizes Ne at different time-points of the experiment. Numerical simulations of asexual haploid Wright-Fisher populations subjected to contrasting genetic drift (Ne ∈ [10, 2000]) and selection (|s| ∈ [0, 0.15]) regimes were used to validate the method proposed. The experiment in closely related pepper host genotypes revealed that viruses experienced a considerable diversity of selection and genetic drift regimes. The resulting variant dynamics were accurately described by Wright-Fisher models. The fitness ranks of the variants were almost identical between host genotypes. By contrast, the dynamics of Ne were highly variable, although a bottleneck was often identified during the systemic movement of the virus. We demonstrated that, for a fixed initial PVY population, virus effective population size is a heritable trait in plants. These findings pave the way for the breeding of plant varieties exposing viruses to stronger genetic drift, thereby slowing virus adaptation.
中文翻译:
估计病毒有效种群的大小和没有中性标记的选择
通过将高通量测序(HTS)与实验进化相结合,我们可以观察到具有生物医学或生态学意义的病原体变异的宿主内部动态。我们研究了马铃薯病毒Y的五个变异体的进化动力学(PVY)在15个双倍单倍体辣椒行中。用相同的病毒变体混合物接种所有植物,并在六个采样日期的每个日期,通过HTS在每个辣椒品系的八株植物中确定变体频率。我们开发了一种在多等位基因赖特-费舍尔模型中估算选择强度和遗传漂移强度的方法,无论这些力是强还是弱,并且在没有中性标记的情况下均适用。此方法需要在独立主机中的几个时间点确定频率变化。参数是每个PVY变体的选择系数以及在实验的不同时间点的四个有效种群大小N e。无性单倍体Wright-Fisher种群受到相反遗传漂移影响的数值模拟(Ñ Ë ∈[10,2000])和选择(|小号|∈[0,0.15])制度被用来验证所提出的方法。在密切相关的辣椒宿主基因型中进行的实验表明,病毒经历了相当多的选择和遗传漂移机制。通过Wright-Fisher模型可以准确地描述所产生的变体动力学。在宿主基因型之间,变体的适应性等级几乎相同。相比之下,N e的动力学尽管在病毒的全身运动过程中经常发现瓶颈,但这些变量的变化很大。我们证明,对于固定的初始PVY种群,病毒有效种群的大小是植物的可遗传性状。这些发现为将病毒暴露于更强的遗传漂移中的植物变种铺平了道路,从而减慢了病毒的适应性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号