当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Four-Component Relativistic Density Functional Calculations of EPR Parameters for Model Complexes of Tungstoenzymes
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1021/acs.jpca.7b08768 Sebastian Gohr 1 , Peter Hrobárik 1, 2 , Martin Kaupp 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1021/acs.jpca.7b08768 Sebastian Gohr 1 , Peter Hrobárik 1, 2 , Martin Kaupp 1
Affiliation
|
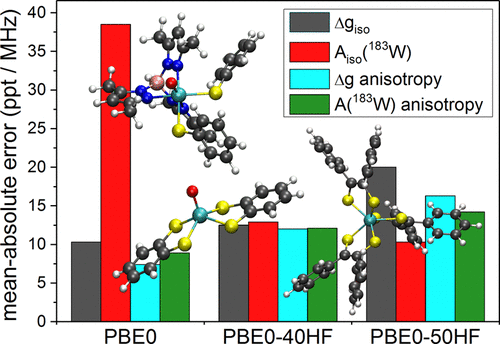
For a closer validation of four-component relativistic DFT methods within the matrix Dirac–Kohn–Sham (mDKS) framework with global hybrid functionals for EPR parameter calculations to be applied in the modeling of tungsten enzymes, we refine a previously suggested protocol for computations on 5d systems. This is done for a series of larger, unsymmetrical W(V) complexes thought to closely resemble enzyme active sites in this oxidation state. Particular focus is placed on complexes with thiolate and dithiolene ligands, along with an evaluation of the influence of different amounts of exact-exchange incorporated in hybrid PBE0-xHF functionals, an implicit solvent model, and structural changes on the computed EPR parameters. Compared to previous work, a slightly modified protocol with different optimal exact-exchange admixtures for electronic g- and hyperfine A-tensors is found to provide the best agreement with experimental EPR data. It will provide the basis for our subsequent tungsten enzyme modeling efforts.
中文翻译:

钨酶模型配合物的EPR参数的四成分相对论密度泛函计算
为了更深入地验证Dirac–Kohn–Sham(mDKS)框架中具有全局混合功能的四组分相对论DFT方法用于EPR参数计算的钨酶模型,我们改进了先前建议的用于计算的协议5d系统。这是针对一系列较大的,不对称的W(V)络合物完成的,这些络合物被认为与该氧化态下的酶活性位点非常相似。特别关注与硫醇盐和二硫代烯配体形成的配合物,以及对掺入杂化PBE0- x中不同数量的精确交换的影响的评估HF功能,隐式溶剂模型和所计算的EPR参数的结构变化。与以前的工作相比,对电子g-和超精细A-张量进行了稍作修改的协议,使用了不同的最佳精确交换混合物,可以与实验性EPR数据提供最佳的一致性。这将为我们后续的钨酶建模工作提供基础。
更新日期:2017-11-17
中文翻译:
钨酶模型配合物的EPR参数的四成分相对论密度泛函计算
为了更深入地验证Dirac–Kohn–Sham(mDKS)框架中具有全局混合功能的四组分相对论DFT方法用于EPR参数计算的钨酶模型,我们改进了先前建议的用于计算的协议5d系统。这是针对一系列较大的,不对称的W(V)络合物完成的,这些络合物被认为与该氧化态下的酶活性位点非常相似。特别关注与硫醇盐和二硫代烯配体形成的配合物,以及对掺入杂化PBE0- x中不同数量的精确交换的影响的评估HF功能,隐式溶剂模型和所计算的EPR参数的结构变化。与以前的工作相比,对电子g-和超精细A-张量进行了稍作修改的协议,使用了不同的最佳精确交换混合物,可以与实验性EPR数据提供最佳的一致性。这将为我们后续的钨酶建模工作提供基础。


























 京公网安备 11010802027423号
京公网安备 11010802027423号