当前位置:
X-MOL 学术
›
Chem. Bio. Drug Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
3D-QSAR, molecular docking, and ONIOM studies on the structure–activity relationships and action mechanism of nitrogen-containing bisphosphonates
Chemical Biology & Drug Design ( IF 3 ) Pub Date : 2017-11-16 00:30:52 , DOI: 10.1111/cbdd.13134 Qing-Zhu Liu 1 , Shan-Shan Wang 1, 2 , Xi Li 1, 2 , Xue-Yu Zhao 1, 2 , Ke Li 1 , Gao-Chao Lv 1 , Ling Qiu 1 , Jian-Guo Lin 1
Chemical Biology & Drug Design ( IF 3 ) Pub Date : 2017-11-16 00:30:52 , DOI: 10.1111/cbdd.13134 Qing-Zhu Liu 1 , Shan-Shan Wang 1, 2 , Xi Li 1, 2 , Xue-Yu Zhao 1, 2 , Ke Li 1 , Gao-Chao Lv 1 , Ling Qiu 1 , Jian-Guo Lin 1
Affiliation
|
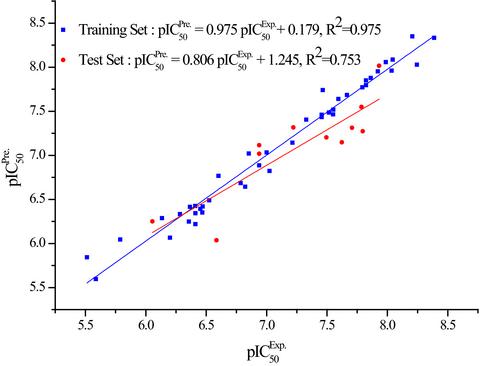
A 3D-QSAR model was constructed for 53 N-BPs with the inhibition activities on hFPPS. A series of novel N-BPs inhibitors were designed and six novel N-BPs inhibitors with better biological activities and higher lipophilicity were screened out. Molecular docking and ONIOM (B3LYP/6-31 + G*:PM6:Amber) calculations showed that the inhibitors bound to the active site of hFPPS via hydrogen-bonding interactions, hydrophobic interactions, and cation-π interactions.
中文翻译:

3D-QSAR,分子对接和ONIOM研究含氮双膦酸酯的结构-活性关系和作用机理
针对53个N-BPs构建了对hFPPS具有抑制活性的3D-QSAR模型。设计了一系列新型的N-BPs抑制剂,并筛选出了六种具有更好的生物活性和更高的亲脂性的新型N-BPs抑制剂。分子对接和ONIOM(B3LYP / 6-31 + G *:PM6:Amber)计算表明,抑制剂通过氢键相互作用,疏水相互作用和阳离子-π相互作用与hFPPS的活性位点结合。
更新日期:2017-11-16
中文翻译:
3D-QSAR,分子对接和ONIOM研究含氮双膦酸酯的结构-活性关系和作用机理
针对53个N-BPs构建了对hFPPS具有抑制活性的3D-QSAR模型。设计了一系列新型的N-BPs抑制剂,并筛选出了六种具有更好的生物活性和更高的亲脂性的新型N-BPs抑制剂。分子对接和ONIOM(B3LYP / 6-31 + G *:PM6:Amber)计算表明,抑制剂通过氢键相互作用,疏水相互作用和阳离子-π相互作用与hFPPS的活性位点结合。

























 京公网安备 11010802027423号
京公网安备 11010802027423号