当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Experimental–Computational Synergy for Selective Pd(II)-Catalyzed C–H Activation of Aryl and Alkyl Groups
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2017-11-08 00:00:00 , DOI: 10.1021/acs.accounts.7b00440 Yun-Fang Yang 1 , Xin Hong 2 , Jin-Quan Yu 3 , K. N. Houk 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2017-11-08 00:00:00 , DOI: 10.1021/acs.accounts.7b00440 Yun-Fang Yang 1 , Xin Hong 2 , Jin-Quan Yu 3 , K. N. Houk 1
Affiliation
|
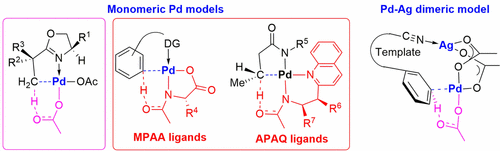
C–H activation and functionalization are on the forefront of modern synthetic chemistry. Imagine if any C–H bond of a molecule could be converted to a C–X bond, where X is the target functionality—this would alter the synthetic blueprints for complex target molecules since it would provide novel disconnections in retrosynthetic analysis. Collaborations between many experimental and computational groups have led to rapid developments of new C–H functionalization methods. Our groups represent an example of this; we were brought together as part of the NSF-supported Center for Selective C–H Functionalization. Many examples of experimental–computational synergy for selective Pd(II)-catalyzed C–H activation of aryl and alkyl groups are described in this Account. We describe computations by the Houk group made in response to experimental stimuli by the Yu group. The first section discusses the experimental and computational investigations of oxazoline-directed stereoselective Pd(II)-catalyzed C(sp3)–H bond activation that occurs through the concerted metalation–deprotonation (CMD) pathway involving a monomeric Pd(II) complex. The second section involves two types of bidentate ligands, mono-N-protected amino acid (MPAA) and acetyl-protected aminoethyl quinoline (APAQ) ligands that promote the C–H activation reactions with the ligand as the internal base. In the MPAA-assisted remote C–H bond activation, the basic dianionic amidate ligand participates in the deprotonation of a specific C–H bond. This mechanism accounts for the improved reactivity and selectivity in C–H activation reactions with MPAA ligands. The chiral APAQ ligands enable asymmetric palladium insertion into prochiral C–H bonds on a single methylene carbon center. The dianionic amidate of the APAQ ligand acts as an intramolecular base to deprotonate the methylene C–H asymmetrically and facilitate chiral Pd–C bond formation. The origins of the dramatic differences of five-membered (relatively inactive) and six-membered (highly active) chelation in β-methylene C(sp3)–H activation reactions by a Pd(II) catalyst were explained with density functional theory (DFT) calculations. This is mainly due to the steric repulsions between the ArF group of the substrate and the quinoline group of the ligand. The steric repulsion between the ArF group of the substrate and the quinoline group of the APAQ ligand destabilizes the five-membered chelate transition structure, increasing the energy of the transition state. The third section discusses a mechanism involving a Pd–Ag heterodimeric complex intermediate in the template-directed, Pd(II)-catalyzed remote meta functionalization of toluene derivatives and benzoic acid derivatives. The nitrile directing group of the template coordinates with Ag while the Pd is placed adjacent to the meta-C–H bond in the transition state, leading to the observed high meta selectivity. The selective activation of remote meta-C–H bonds at various distances can be achieved by tuning the template. The dual role of AgOAc as both an oxidant and part of the heteronuclear active species in the mechanism involving PdAg(OAc)3 was determined by DFT calculations and is in accord with literature information about complexes. For the systems discussed in these three sections, the similarity is that they all proceed via the CMD mechanism. The differences lie in the proton acceptors and the active Pd species. Common CMD involves a monomeric Pd mechanism with acetate as the proton acceptor. Both MPAA and APAQ ligands react via monomeric Pd mechanisms with a ligand moiety (the amidate oxygen) as the proton acceptor. Nitrile-containing template-mediated meta-C–H activations proceed via a Pd–Ag heterodimeric mechanism, still with acetate as the proton acceptor. The interaction between our two groups, experts in experiment and computation, and the discoveries made possible by that interplay are highlighted in this Account.
中文翻译:

选择性Pd(II)催化芳基和烷基的C–H活化的实验-计算协同作用
C–H活化和功能化处于现代合成化学的最前沿。想象一下,如果某个分子的任何C–H键可以转换为C–X键,其中X是目标官能团,这将改变复杂目标分子的合成蓝图,因为它将在逆合成分析中提供新颖的分离。许多实验和计算小组之间的合作导致了新的C–H功能化方法的迅速发展。我们的小组就是一个例子。我们是NSF支持的选择性C–H功能化中心的一部分。此帐户中描述了许多用于选择性Pd(II)催化芳基和烷基的C–H活化的实验-计算协同作用的例子。我们描述了Houk小组针对Yu小组的实验刺激所做的计算。第一部分讨论了恶唑啉定向的立体选择性Pd(II)催化的C(sp3)– H键激活通过涉及单体Pd(II)配合物的协同金属化-去质子化(CMD)途径发生。第二部分涉及两种类型的双齿配体,单氮原子保护的氨基酸(MPAA)和乙酰基保护的氨基乙基喹啉(APAQ)配体,它们以配体作为内部碱基促进C–H活化反应。在MPAA辅助的远程C–H键激活中,碱性双阴离子ionic酸酯配体参与了特定C–H键的去质子化。这种机制解释了与MPAA配体在C–H活化反应中提高的反应性和选择性。手性APAQ配体可将不对称钯插入单个亚甲基碳中心的前手性CH键中。APAQ配体的双阴离子a酸盐可作为分子内碱基,使亚甲基CH不对称地去质子化并促进手性Pd-C键的形成。β-亚甲基C(sp)中五元(相对不活跃)和六元(高度活跃)螯合作用的戏剧性差异的起因3)-Pd(II)催化剂的H活化反应用密度泛函理论(DFT)计算进行了解释。这主要是由于底物的Ar F基与配体的喹啉基之间的空间排斥。底物的Ar F基团与APAQ配体的喹啉基团之间的空间排斥作用会破坏五元螯合物过渡结构的稳定性,从而增加过渡态的能量。第三部分讨论了一种机制,该机制涉及在模板导向的Pd(II)催化的甲苯衍生物和苯甲酸衍生物的远位元官能化中涉及Pd-Ag异二聚体复杂中间体。当Pd放置在与模板相邻的位置时,模板的腈引导基团与Ag配合。处于过渡态的meta -C–H键,导致观察到的高meta选择性。可以通过调节模板来实现在不同距离处选择性地激活远程meta -C–H键。在涉及PdAg(OAc)3的机制中,AgOAc作为氧化剂和部分异核活性物质的双重作用由DFT计算确定,并且与有关配合物的文献信息相符。对于这三部分中讨论的系统,相似之处在于它们都是通过CMD机制进行的。差异在于质子受体和活性Pd物种。常见的CMD涉及单体Pd机理,其中乙酸盐是质子受体。MPAA和APAQ配体都通过单体Pd机理与作为质子受体的配体部分(酰胺酸氧)反应。含腈的模板介导的间位C-H活化是通过Pd-Ag异二聚机制进行的,但仍以乙酸盐为质子受体。该帐户重点介绍了我们这两个小组(实验和计算专家)之间的互动,以及通过这种相互作用而实现的发现。
更新日期:2017-11-08
中文翻译:
选择性Pd(II)催化芳基和烷基的C–H活化的实验-计算协同作用
C–H活化和功能化处于现代合成化学的最前沿。想象一下,如果某个分子的任何C–H键可以转换为C–X键,其中X是目标官能团,这将改变复杂目标分子的合成蓝图,因为它将在逆合成分析中提供新颖的分离。许多实验和计算小组之间的合作导致了新的C–H功能化方法的迅速发展。我们的小组就是一个例子。我们是NSF支持的选择性C–H功能化中心的一部分。此帐户中描述了许多用于选择性Pd(II)催化芳基和烷基的C–H活化的实验-计算协同作用的例子。我们描述了Houk小组针对Yu小组的实验刺激所做的计算。第一部分讨论了恶唑啉定向的立体选择性Pd(II)催化的C(sp3)– H键激活通过涉及单体Pd(II)配合物的协同金属化-去质子化(CMD)途径发生。第二部分涉及两种类型的双齿配体,单氮原子保护的氨基酸(MPAA)和乙酰基保护的氨基乙基喹啉(APAQ)配体,它们以配体作为内部碱基促进C–H活化反应。在MPAA辅助的远程C–H键激活中,碱性双阴离子ionic酸酯配体参与了特定C–H键的去质子化。这种机制解释了与MPAA配体在C–H活化反应中提高的反应性和选择性。手性APAQ配体可将不对称钯插入单个亚甲基碳中心的前手性CH键中。APAQ配体的双阴离子a酸盐可作为分子内碱基,使亚甲基CH不对称地去质子化并促进手性Pd-C键的形成。β-亚甲基C(sp)中五元(相对不活跃)和六元(高度活跃)螯合作用的戏剧性差异的起因3)-Pd(II)催化剂的H活化反应用密度泛函理论(DFT)计算进行了解释。这主要是由于底物的Ar F基与配体的喹啉基之间的空间排斥。底物的Ar F基团与APAQ配体的喹啉基团之间的空间排斥作用会破坏五元螯合物过渡结构的稳定性,从而增加过渡态的能量。第三部分讨论了一种机制,该机制涉及在模板导向的Pd(II)催化的甲苯衍生物和苯甲酸衍生物的远位元官能化中涉及Pd-Ag异二聚体复杂中间体。当Pd放置在与模板相邻的位置时,模板的腈引导基团与Ag配合。处于过渡态的meta -C–H键,导致观察到的高meta选择性。可以通过调节模板来实现在不同距离处选择性地激活远程meta -C–H键。在涉及PdAg(OAc)3的机制中,AgOAc作为氧化剂和部分异核活性物质的双重作用由DFT计算确定,并且与有关配合物的文献信息相符。对于这三部分中讨论的系统,相似之处在于它们都是通过CMD机制进行的。差异在于质子受体和活性Pd物种。常见的CMD涉及单体Pd机理,其中乙酸盐是质子受体。MPAA和APAQ配体都通过单体Pd机理与作为质子受体的配体部分(酰胺酸氧)反应。含腈的模板介导的间位C-H活化是通过Pd-Ag异二聚机制进行的,但仍以乙酸盐为质子受体。该帐户重点介绍了我们这两个小组(实验和计算专家)之间的互动,以及通过这种相互作用而实现的发现。

























 京公网安备 11010802027423号
京公网安备 11010802027423号