当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Twisting and Tilting 1,1′-Bis(dialkylphosphino)ferrocene Bound to Low Valent Tricarbonylmaganese(I to −I)
Inorganic Chemistry ( IF 4.6 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1021/acs.inorgchem.7b02627 K. Michael Schäfer 1 , Leonie Reinders 1 , Jan Fiedler 2 , Mark R. Ringenberg 1
Inorganic Chemistry ( IF 4.6 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1021/acs.inorgchem.7b02627 K. Michael Schäfer 1 , Leonie Reinders 1 , Jan Fiedler 2 , Mark R. Ringenberg 1
Affiliation
|
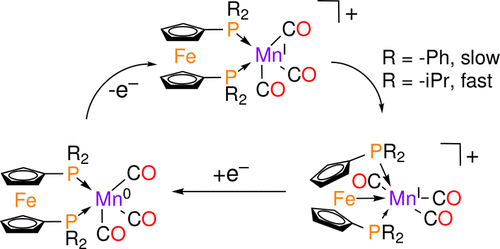
Recently we had reported the noninnocent behavior of 1,1′-bis(diphenylphosphino)ferrocene (dppf) in Fe(CO)3dppf [Ringenberg et al., Inorg. Chem., 2017, 56, 7501]. Moving to the left in the periodic table, HMn(CO)3(dRpf) where dRpf = dppf (1H) and 1,1′-bis(diisopropylphosphino)ferrocene (dippf) (2H) were synthesized. The hydride ligand was removed by protonation with [(Et2O)2H][B(ArF)4] ([B(ArF)4]− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate), resulting in the rapid evolution of H2 followed by the formation of an Fe→Mn interaction. The reaction mechanism was determined by in situ IR experiments which show that directly following protonation both [1]+ and [2]+ offer an open manganese coordination site that allows for the formation of an intramolecular Fe→Mn dative bond. This process is significantly faster for [2]+ than for [1]+. The reduction chemistry as studied by cyclic voltammetry (CV) reveals that both complexes change from a distorted octahedral coordination with an Fe→Mn interaction to an open square-pyramidal configuration which is more stable for [1]0 than [2]0. Reoxidation of this square-pyramidal species proceeds more reversibly for 2 versus 1 due to the faster ferrocene ligand reorganization. The electrochemical mechanism was studied by in situ spectroscopic techniques, e.g., IR, UV–vis–NIR (near IR), and EPR spectroelectrochemistry (SEC) as well as by CV simulation. The new complexes described offer an exciting platform for the development of electrocatalysts for the reduction of CO2 to CO, or for proton reduction (2H+ + 2e– → H2).
中文翻译:

将1,1'-双(二烷基膦基)二茂铁扭转并倾斜成低价三羰基锰(I至-I)
最近,我们已经报道了1,1'-双(二苯基膦基)二茂铁(dppf)在Fe(CO)3 dppf中的无毒行为[Ringenberg等。,Inorg。化学 ,2017,56,7501]。移至元素周期表的左侧,合成了HMn(CO)3(dRpf),其中dRpf = dppf(1H)和1,1'-双(二异丙基膦基)二茂铁(dippf)(2H)。通过[[Et 2 O)2 H] [B(Ar F)4 ]([[B(Ar F)4 ] -=四[3,5-双(三氟甲基)苯基]硼酸酯),导致H 2快速放出,随后形成Fe→Mn相互作用。反应机理是通过原位红外实验确定的,该实验表明质子化后[ 1 ] +和[ 2 ] +都提供了一个开放的锰配位位点,从而可以形成分子内的Fe→Mn配位键。对于[ 2 ] +,此过程明显快于[ 1 ] +。通过循环伏安法(CV)进行的还原化学分析表明,两种配合物都从具有Fe→Mn相互作用的扭曲的八面体配位转变为[ 1 ] 0比[ 2 ] 0更稳定的开方角锥体结构。这四方锥物种的再氧化进行更可逆地为2对比1由于更快二茂铁配体重组。电化学机理进行了原位研究光谱技术,例如IR,UV-vis-NIR(近IR),EPR光谱电化学(SEC)以及CV模拟。所述的新络合物为开发用于将CO 2还原为CO或质子还原(2H + + 2e – →H 2)的电催化剂提供了令人兴奋的平台。
更新日期:2017-11-16
中文翻译:
将1,1'-双(二烷基膦基)二茂铁扭转并倾斜成低价三羰基锰(I至-I)
最近,我们已经报道了1,1'-双(二苯基膦基)二茂铁(dppf)在Fe(CO)3 dppf中的无毒行为[Ringenberg等。,Inorg。化学 ,2017,56,7501]。移至元素周期表的左侧,合成了HMn(CO)3(dRpf),其中dRpf = dppf(1H)和1,1'-双(二异丙基膦基)二茂铁(dippf)(2H)。通过[[Et 2 O)2 H] [B(Ar F)4 ]([[B(Ar F)4 ] -=四[3,5-双(三氟甲基)苯基]硼酸酯),导致H 2快速放出,随后形成Fe→Mn相互作用。反应机理是通过原位红外实验确定的,该实验表明质子化后[ 1 ] +和[ 2 ] +都提供了一个开放的锰配位位点,从而可以形成分子内的Fe→Mn配位键。对于[ 2 ] +,此过程明显快于[ 1 ] +。通过循环伏安法(CV)进行的还原化学分析表明,两种配合物都从具有Fe→Mn相互作用的扭曲的八面体配位转变为[ 1 ] 0比[ 2 ] 0更稳定的开方角锥体结构。这四方锥物种的再氧化进行更可逆地为2对比1由于更快二茂铁配体重组。电化学机理进行了原位研究光谱技术,例如IR,UV-vis-NIR(近IR),EPR光谱电化学(SEC)以及CV模拟。所述的新络合物为开发用于将CO 2还原为CO或质子还原(2H + + 2e – →H 2)的电催化剂提供了令人兴奋的平台。


























 京公网安备 11010802027423号
京公网安备 11010802027423号