当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Influence of Hubbard U Parameter in Simulating Adsorption and Reactivity on CuO: Combined Theoretical and Experimental Study
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2017-09-25 00:00:00 , DOI: 10.1021/acs.jpcc.7b05385 Kartavya Bhola 1 , Jithin John Varghese 2 , Liu Dapeng 3 , Yan Liu 3 , Samir H. Mushrif 1, 2
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2017-09-25 00:00:00 , DOI: 10.1021/acs.jpcc.7b05385 Kartavya Bhola 1 , Jithin John Varghese 2 , Liu Dapeng 3 , Yan Liu 3 , Samir H. Mushrif 1, 2
Affiliation
|
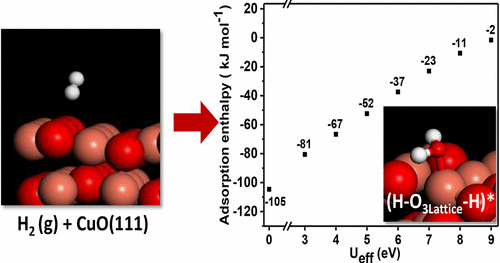
Transition metal oxides are an important class of catalytic materials widely used in the chemical manufacturing and processing industry, owing to their low cost, high surface area, low toxicity, and easily tunable surface and structural properties. For these strongly correlated transition metal oxides, standard approximations in the density functional theory (DFT) exchange-correlation functional fail to describe the electron localization accurately due to the intrinsic errors arising from electron self-interactions. DFT+U method is a widely used extension of DFT, where the Hubbard U term is an onsite potential which puts a penalty on electron delocalization, successfully describing such systems at only slightly higher computational cost than standard DFT methods. The U-value is usually chosen based on its accuracy in reproducing bulk properties like lattice parameters and band structure. However, chemical reactions on transition metal oxide surfaces involve complex surface–adsorbate interactions, and using the bulk properties based U-values in a locally changing surface environment may not describe reaction energetics correctly. Hence, in the current DFT+U benchmarking work, using CuO as a model transition metal oxide, we perform DFT+U calculations to investigate the dissociative chemisorption of H2 on it. It is observed that the U-value impacts computed adsorption enthalpies by over 100 kJ mol–1. The DFT+U calculated adsorption enthalpy is compared with the experimental adsorption enthalpy, and equilibrium adsorption configurations are confirmed using infrared analysis. We reveal that the commonly used U-value of 7 eV (fitted against CuO bulk properties) overestimates the adsorption enthalpy by 20–40 kJ mol–1. The U-value between 4.5 and 5.5 eV correctly predicts the adsorption of H2 on CuO. The DFT+U benchmarking procedure elucidated in this article, encapsulates surface–adsorbate interactions, surface reactivity, and the dynamic surface reaction environment and, thus, provides an appropriate U-value to be used to model reactions on metal oxide surfaces.
中文翻译:

Hubbard U参数在模拟吸附和反应性上对CuO的影响:理论和实验相结合
过渡金属氧化物由于其低成本,高表面积,低毒性以及易于调节的表面和结构特性而成为在化学制造和加工工业中广泛使用的重要催化材料类别。对于这些高度相关的过渡金属氧化物,由于电子自相互作用引起的固有误差,在密度泛函理论(DFT)交换-相关函数中的标准近似无法准确描述电子局部化。DFT + U方法是DFT的一种广泛使用的扩展,其中Hubbard U项是一种现场电势,对电子离域产生了不利影响,以比标准DFT方法略高的计算成本成功地描述了此类系统。通常根据其在重现诸如晶格参数和能带结构之类的整体性质时的准确性来选择U值。但是,过渡金属氧化物表面上的化学反应涉及复杂的表面-吸附物相互作用,在局部变化的表面环境中使用基于整体性质的U值可能无法正确描述反应能。因此,在当前的DFT + U基准测试工作中,使用CuO作为过渡金属氧化物模型,我们执行DFT + U计算以研究H的解离化学吸附2就可以了。可以看出,U值影响了超过100 kJ mol –1的计算的吸附焓。将DFT + U计算的吸附焓与实验吸附焓进行比较,并使用红外分析确定平衡吸附构型。我们发现,常用的7 eV的U值(适合CuO的整体性质)高估了20–40 kJ mol –1的吸附焓。U值介于4.5和5.5 eV之间可正确预测H 2的吸附在CuO上。本文阐述的DFT + U基准测试程序封装了表面与被吸附物的相互作用,表面反应性和动态表面反应环境,因此提供了用于模拟金属氧化物表面反应的适当U值。
更新日期:2017-09-26
中文翻译:
Hubbard U参数在模拟吸附和反应性上对CuO的影响:理论和实验相结合
过渡金属氧化物由于其低成本,高表面积,低毒性以及易于调节的表面和结构特性而成为在化学制造和加工工业中广泛使用的重要催化材料类别。对于这些高度相关的过渡金属氧化物,由于电子自相互作用引起的固有误差,在密度泛函理论(DFT)交换-相关函数中的标准近似无法准确描述电子局部化。DFT + U方法是DFT的一种广泛使用的扩展,其中Hubbard U项是一种现场电势,对电子离域产生了不利影响,以比标准DFT方法略高的计算成本成功地描述了此类系统。通常根据其在重现诸如晶格参数和能带结构之类的整体性质时的准确性来选择U值。但是,过渡金属氧化物表面上的化学反应涉及复杂的表面-吸附物相互作用,在局部变化的表面环境中使用基于整体性质的U值可能无法正确描述反应能。因此,在当前的DFT + U基准测试工作中,使用CuO作为过渡金属氧化物模型,我们执行DFT + U计算以研究H的解离化学吸附2就可以了。可以看出,U值影响了超过100 kJ mol –1的计算的吸附焓。将DFT + U计算的吸附焓与实验吸附焓进行比较,并使用红外分析确定平衡吸附构型。我们发现,常用的7 eV的U值(适合CuO的整体性质)高估了20–40 kJ mol –1的吸附焓。U值介于4.5和5.5 eV之间可正确预测H 2的吸附在CuO上。本文阐述的DFT + U基准测试程序封装了表面与被吸附物的相互作用,表面反应性和动态表面反应环境,因此提供了用于模拟金属氧化物表面反应的适当U值。


























 京公网安备 11010802027423号
京公网安备 11010802027423号