当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Voltage Dependence of Molecule–Electrode Coupling in Biased Molecular Junctions
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2017-09-20 00:00:00 , DOI: 10.1021/acs.jpcc.7b05567 Zhen-Fei Liu 1, 2 , Jeffrey B. Neaton 1, 2, 3
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2017-09-20 00:00:00 , DOI: 10.1021/acs.jpcc.7b05567 Zhen-Fei Liu 1, 2 , Jeffrey B. Neaton 1, 2, 3
Affiliation
|
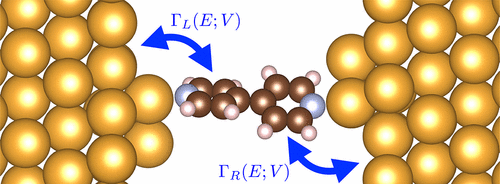
Biased interfaces between individual molecules and metallic electrodes are central in many nanoscale devices. A key quantity that governs the nature of the metal–molecule interface is the degree of hybridization between frontier orbitals of the molecule and electrode continuum states. This hybridization leads to the broadening of molecular resonances, a measure of the electronic coupling to the surface. In this work, we present detailed ab initio calculations of the evolution of this electronic coupling for selected frontier orbitals with bias voltage in molecular junctions, nanoscale devices where metal–molecule interface effects are dominant. We focus on symmetric and asymmetric junctions based on an experimentally well-studied system, Au–bipyridine–Au, as examples of our method. The nature of the bias-dependent coupling that emerges from our ab initio calculations provides new physical insight into experimentally achievable systems and is a marked quantitative improvement over simple Lorentzian models often used in interpreting device measurements. Beyond molecular junctions, our approach can be straightforwardly generalized to other molecule–metal interfaces.
中文翻译:

偏置分子结中分子-电极耦合的电压依赖性
在许多纳米级设备中,单个分子与金属电极之间的偏向界面很重要。决定金属-分子界面性质的关键因素是分子前沿轨道与电极连续态之间的杂合度。这种杂交导致分子共振的扩大,这是与表面电子耦合的一种量度。在这项工作中,我们对分子边界,金属-分子界面效应占主导地位的纳米级器件中具有偏置电压的选定前沿轨道的这种电子耦合的演化过程进行了详细的从头算计算。我们重点研究基于实验研究良好的系统Au-bipyridine-Au的对称和不对称结,作为我们方法的示例。从头算计算得出的依赖于偏差的耦合的性质为实验上可实现的系统提供了新的物理见解,并且相对于通常用于解释设备测量值的简单洛伦兹模型具有明显的定量改进。除了分子结,我们的方法还可以直接推广到其他分子-金属界面。
更新日期:2017-09-20
中文翻译:
偏置分子结中分子-电极耦合的电压依赖性
在许多纳米级设备中,单个分子与金属电极之间的偏向界面很重要。决定金属-分子界面性质的关键因素是分子前沿轨道与电极连续态之间的杂合度。这种杂交导致分子共振的扩大,这是与表面电子耦合的一种量度。在这项工作中,我们对分子边界,金属-分子界面效应占主导地位的纳米级器件中具有偏置电压的选定前沿轨道的这种电子耦合的演化过程进行了详细的从头算计算。我们重点研究基于实验研究良好的系统Au-bipyridine-Au的对称和不对称结,作为我们方法的示例。从头算计算得出的依赖于偏差的耦合的性质为实验上可实现的系统提供了新的物理见解,并且相对于通常用于解释设备测量值的简单洛伦兹模型具有明显的定量改进。除了分子结,我们的方法还可以直接推广到其他分子-金属界面。

























 京公网安备 11010802027423号
京公网安备 11010802027423号