Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Redefining the Protein–Protein Interface: Coarse Graining and Combinatorics for an Improved Understanding of Amino Acid Contributions to the Protein–Protein Binding Affinity
Langmuir ( IF 3.9 ) Pub Date : 2017-09-12 00:00:00 , DOI: 10.1021/acs.langmuir.7b02438 Josh K. Smith 1 , Shaoyi Jiang 1 , Jim Pfaendtner 1
Langmuir ( IF 3.9 ) Pub Date : 2017-09-12 00:00:00 , DOI: 10.1021/acs.langmuir.7b02438 Josh K. Smith 1 , Shaoyi Jiang 1 , Jim Pfaendtner 1
Affiliation
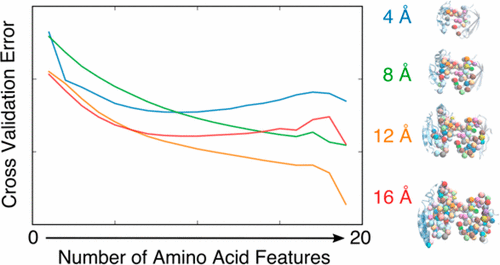
|
The ability to intervene in biological pathways has for decades been limited by the lack of a quantitative description of protein–protein interactions (PPIs). Herein we generate and compare millions of simple PPI models for insight into the mechanisms of specific recognition and binding. We use a coarse-grained approach whereby amino acids are counted in the interface, and these counts are used as binding affinity predictors. We perform lasso regression, a modern regression technique aimed at interpretability, with every possible amino acid combination (over 106 unique feature sets) to select only those amino acid predictors that provide more information than noise. This approach circumvents arbitrary binning and assumptions about the binding environment that obscure other binding affinity models. Aggregated analysis of these models trained at various interfacial cutoff distances informs the roles of specific amino acids in different binding contexts. We find that a simple amino acid count model outperforms detailed intermolecular contact and binned residue type models. We identify the prevalence of serine, glycine, and tryptophan in the interface as particularly important for predicting binding affinity across a range of distance cutoffs. Although current sample size limitations prevent a robust consensus model for binding affinity prediction, our approach underscores the relevance of a residue-based description of the protein–protein interface to increase our understanding of specific interactions.
中文翻译:

重新定义蛋白质-蛋白质界面:粗粒和组合用于更好地了解氨基酸对蛋白质-蛋白质结合亲和力的作用
几十年来,由于缺乏蛋白质间相互作用(PPI)的定量描述,干预生物途径的能力受到限制。在这里,我们生成并比较了数百万个简单的PPI模型,以洞悉特定的识别和绑定机制。我们使用一种粗粒度方法,其中在界面中对氨基酸进行计数,并将这些计数用作结合亲和力预测因子。我们执行套索回归,这是一种针对可解释性的现代回归技术,包含每种可能的氨基酸组合(超过10 6独特的功能集),仅选择那些能提供比噪音更多信息的氨基酸预测因子。这种方法规避了任意装仓和关于使其他绑定亲和力模型模糊的绑定环境的假设。在各种界面截止距离下训练的这些模型的综合分析,揭示了特定氨基酸在不同结合环境中的作用。我们发现简单的氨基酸计数模型优于详细的分子间接触和装仓残基类型模型。我们确定界面中丝氨酸,甘氨酸和色氨酸的普遍性对于预测跨距离范围的结合亲和力尤为重要。尽管当前的样本量限制无法为结合亲和力预测提供可靠的共识模型,
更新日期:2017-09-12
中文翻译:
重新定义蛋白质-蛋白质界面:粗粒和组合用于更好地了解氨基酸对蛋白质-蛋白质结合亲和力的作用
几十年来,由于缺乏蛋白质间相互作用(PPI)的定量描述,干预生物途径的能力受到限制。在这里,我们生成并比较了数百万个简单的PPI模型,以洞悉特定的识别和绑定机制。我们使用一种粗粒度方法,其中在界面中对氨基酸进行计数,并将这些计数用作结合亲和力预测因子。我们执行套索回归,这是一种针对可解释性的现代回归技术,包含每种可能的氨基酸组合(超过10 6独特的功能集),仅选择那些能提供比噪音更多信息的氨基酸预测因子。这种方法规避了任意装仓和关于使其他绑定亲和力模型模糊的绑定环境的假设。在各种界面截止距离下训练的这些模型的综合分析,揭示了特定氨基酸在不同结合环境中的作用。我们发现简单的氨基酸计数模型优于详细的分子间接触和装仓残基类型模型。我们确定界面中丝氨酸,甘氨酸和色氨酸的普遍性对于预测跨距离范围的结合亲和力尤为重要。尽管当前的样本量限制无法为结合亲和力预测提供可靠的共识模型,


























 京公网安备 11010802027423号
京公网安备 11010802027423号