当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Substrate-Specific Screening for Mutational Hotspots Using Biased Molecular Dynamics Simulations
ACS Catalysis ( IF 12.9 ) Pub Date : 2017-09-11 00:00:00 , DOI: 10.1021/acscatal.7b02634 Maximilian C. C. J. C. Ebert 1, 2 , Joaquin Guzman Espinola 1, 2 , Guillaume Lamoureux 2, 3 , Joelle N. Pelletier 1, 2, 4
ACS Catalysis ( IF 12.9 ) Pub Date : 2017-09-11 00:00:00 , DOI: 10.1021/acscatal.7b02634 Maximilian C. C. J. C. Ebert 1, 2 , Joaquin Guzman Espinola 1, 2 , Guillaume Lamoureux 2, 3 , Joelle N. Pelletier 1, 2, 4
Affiliation
|
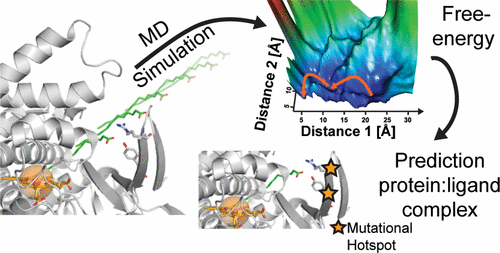
Prediction of substrate-specific mutational hotspots for enzyme engineering is a complex and computationally intensive task. This becomes particularly challenging when the available crystal structures have no ligand, bind a distant homologue of the desired substrate, or hold the ligand in a nonproductive conformation. To address that shortcoming, we present a combined molecular dynamics simulation and molecular docking protocol to predict the conformation of catalytically relevant enzyme–ligand complexes even in the absence of a ligand-bound structure. We applied the adaptive biasing force method to predict the ligand-specific path of diffusion of a fatty acid substrate from the bulk media into the active site of cytochrome P450 CYP102A1 (BM3). Starting with a ligand-free crystal structure, we successfully identified all residues known to be involved in palmitic acid binding to BM3. The binding trajectory also revealed a yet unknown binding residue, Q73, which we confirmed experimentally. Building the free-energy landscape illustrates that, similar to human cytochrome P450s, binding is multistep and does not follow simple Michaelis–Menten kinetics. We confirmed the robustness of the method using a structurally distinct substrate, the small aromatic indole. We then applied the predicted BM3:palmitate complex to molecular docking of a library of 29 palmitate analogues. This produced catalytically relevant binding poses for the entire library, while docking directly into ligand-free and ligand-bound crystal structures gave poor results. This fast and simple computational method is broadly applicable for predicting binding hotspots in a substrate-specific manner and has the potential to drastically reduce the experimental screening effort to tailor an enzyme to substrates of interest.
中文翻译:

使用偏向分子动力学模拟对突变热点进行底物特异性筛选
酶工程学的底物特异性突变热点的预测是一项复杂且计算量大的任务。当可用的晶体结构没有配体,结合所需底物的遥远同源物或使配体保持非生产性构型时,这尤其具有挑战性。为了解决该缺点,我们提出了一种结合分子动力学模拟和分子对接方案的方法,即使在没有配体结合的结构的情况下,也可以预测具有催化作用的酶-配体复合物的构象。我们应用了自适应偏压力方法来预测脂肪酸底物从整体培养基向细胞色素P450 CYP102A1(BM3)的活性位点扩散的配体特异性路径。从无配体的晶体结构开始 我们成功鉴定了所有已知的棕榈酸与BM3结合的残基。结合轨迹还揭示了一个未知的结合残基Q73,我们通过实验证实了这一点。建立自由能态势表明,与人类细胞色素P450相似,结合是多步骤的,并不遵循简单的米利斯-门腾动力学。我们确认了使用结构独特的底物小芳族吲哚的方法的鲁棒性。然后,我们将预测的BM3:棕榈酸酯复合物应用于29个棕榈酸酯类似物库的分子对接。这为整个文库产生了催化相关的结合姿势,而直接对接成无配体和与配体结合的晶体结构却产生了不良结果。
更新日期:2017-09-11
中文翻译:
使用偏向分子动力学模拟对突变热点进行底物特异性筛选
酶工程学的底物特异性突变热点的预测是一项复杂且计算量大的任务。当可用的晶体结构没有配体,结合所需底物的遥远同源物或使配体保持非生产性构型时,这尤其具有挑战性。为了解决该缺点,我们提出了一种结合分子动力学模拟和分子对接方案的方法,即使在没有配体结合的结构的情况下,也可以预测具有催化作用的酶-配体复合物的构象。我们应用了自适应偏压力方法来预测脂肪酸底物从整体培养基向细胞色素P450 CYP102A1(BM3)的活性位点扩散的配体特异性路径。从无配体的晶体结构开始 我们成功鉴定了所有已知的棕榈酸与BM3结合的残基。结合轨迹还揭示了一个未知的结合残基Q73,我们通过实验证实了这一点。建立自由能态势表明,与人类细胞色素P450相似,结合是多步骤的,并不遵循简单的米利斯-门腾动力学。我们确认了使用结构独特的底物小芳族吲哚的方法的鲁棒性。然后,我们将预测的BM3:棕榈酸酯复合物应用于29个棕榈酸酯类似物库的分子对接。这为整个文库产生了催化相关的结合姿势,而直接对接成无配体和与配体结合的晶体结构却产生了不良结果。


























 京公网安备 11010802027423号
京公网安备 11010802027423号