当前位置:
X-MOL 学术
›
Organometallics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Ligand Exchange on and Allylic C–H Activation by Iron(0) Fragments: π-Complexes, Allyliron Species, and Metallacycles
Organometallics ( IF 2.8 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1021/acs.organomet.7b00571 Alicia Casitas 1 , Helga Krause 1 , Sigrid Lutz 1 , Richard Goddard 1 , Eckhard Bill 2 , Alois Fürstner 1
Organometallics ( IF 2.8 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1021/acs.organomet.7b00571 Alicia Casitas 1 , Helga Krause 1 , Sigrid Lutz 1 , Richard Goddard 1 , Eckhard Bill 2 , Alois Fürstner 1
Affiliation
|
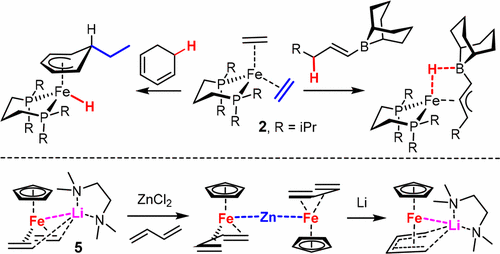
The complexes [(dippp)Fe(C2H4)2] (2) and [CpFe(C2H4)2][Li·(tmeda)] (5) both contain a formally zerovalent iron center but exhibit markedly different catalytic properties. Whereas 5 is able to induce a broad range of cycloisomerization and cycloaddition reactions, 2 is so far basically limited to cyclotrimerizations of alkynes and nitriles. Investigations into the behaviors of both complex vis-à-vis unsaturated substrates provided insights into the likely origins of this distinct behavior. Thus, ordinary terminal or internal alkenes were found not to replace the ligated ethylene units in 2, whereas the stronger π-acceptor ligands 1,5-cyclooctadiene, 2-norbornene, and tolane afforded the corresponding π-complexes 8, 9, 10, and 13. A cyclopropene derivative engaged in oxidative cyclization with formation of the corresponding metallacycle 12. Allyl-9-BBN or alkenyl-9-BBN derivatives succumbed to allylic C–H activation with formation of the unorthodox allyliron complexes 25 and 27 featuring a bridging hydride ligand between the iron and the boron atoms. Along the same line, 1,3-dienes bind well to 2 but undergo spontaneous activation if allylic C–H bonds are present; the resulting hydride is transferred to a residual ethylene ligand, as manifest in the formation of the cyclopentadienyl ethyl complex 22. The same elementary steps surface in a remarkable reaction cascade comprising two consecutive C–H activation reactions and a stereoselective C–C bond formation, which ultimately provides the substituted cyclohexadienyl complexes 20 and 23. In contrast, the heterobimetallic complex 5 neither induces allylic C–H activation nor binds 1,3-butadiene under conditions where it proved catalytically active. The targeted butadiene complex 34 had to be made by an indirect route and is distinguished by a noteworthy “flyover” constitution. Therefore, we conclude that the known cycloaddition and cycloisomerization reactions catalyzed by 5 do not commence at a 1,3-diene motif but require an enyne entity as starter unit.
中文翻译:

铁(0)片段上的配体交换和烯丙基CH活化:π-络合物,烯丙基铁物种和金属环
配合物[(dippp)Fe(C 2 H 4)2 ](2)和[CpFe(C 2 H 4)2 ] [Li·(tmeda)](5)都含有形式上为零价的铁中心,但表现出明显不同催化性能。尽管5能够诱导广泛的环异构化和环加成反应,2到目前为止,基本上仅限于炔烃和腈的环三聚。对两种复杂的相对于不饱和基材的行为的研究提供了对这种独特行为的可能起源的见解。因此,发现普通末端或内部烯烃不更换连接的乙烯单元2,而较强的π -受体配体1,5-环辛二烯,2-降冰片烯,和二苯基乙炔,得到相应的π络合物8,9,10,和13。参与氧化环化并形成相应金属环12的环丙烯衍生物。烯丙基-9-BBN或烯基-9-BBN衍生物屈服于烯丙基CH活化,并形成了非正统的烯丙基铁配合物25和27,其特征是铁和硼原子之间具有桥联氢化物配体。同样地,如果存在烯丙基CH键,则1,3-二烯与2结合良好,但会自发活化。如形成环戊二烯基乙基络合物22所示,将所得的氢化物转移至残留的乙烯配体。相同的基本步骤在一个显着的反应级联中浮出水面,该级联反应包括两个连续的C–H活化反应和立体选择性C–C键形成,最终形成了取代的环己二烯基络合物20和23。相反,在证明其具有催化活性的条件下,杂双金属配合物5既不诱导烯丙基CH活化,也不与1,3-丁二烯结合。靶向丁二烯配合物34必须通过间接途径制备,并以值得注意的“立交”结构为特征。因此,我们得出结论,已知由5催化的环加成和环异构化反应并非始于1,3-二烯基序,而是需要一个烯炔实体作为起始单元。
更新日期:2018-02-02
中文翻译:
铁(0)片段上的配体交换和烯丙基CH活化:π-络合物,烯丙基铁物种和金属环
配合物[(dippp)Fe(C 2 H 4)2 ](2)和[CpFe(C 2 H 4)2 ] [Li·(tmeda)](5)都含有形式上为零价的铁中心,但表现出明显不同催化性能。尽管5能够诱导广泛的环异构化和环加成反应,2到目前为止,基本上仅限于炔烃和腈的环三聚。对两种复杂的相对于不饱和基材的行为的研究提供了对这种独特行为的可能起源的见解。因此,发现普通末端或内部烯烃不更换连接的乙烯单元2,而较强的π -受体配体1,5-环辛二烯,2-降冰片烯,和二苯基乙炔,得到相应的π络合物8,9,10,和13。参与氧化环化并形成相应金属环12的环丙烯衍生物。烯丙基-9-BBN或烯基-9-BBN衍生物屈服于烯丙基CH活化,并形成了非正统的烯丙基铁配合物25和27,其特征是铁和硼原子之间具有桥联氢化物配体。同样地,如果存在烯丙基CH键,则1,3-二烯与2结合良好,但会自发活化。如形成环戊二烯基乙基络合物22所示,将所得的氢化物转移至残留的乙烯配体。相同的基本步骤在一个显着的反应级联中浮出水面,该级联反应包括两个连续的C–H活化反应和立体选择性C–C键形成,最终形成了取代的环己二烯基络合物20和23。相反,在证明其具有催化活性的条件下,杂双金属配合物5既不诱导烯丙基CH活化,也不与1,3-丁二烯结合。靶向丁二烯配合物34必须通过间接途径制备,并以值得注意的“立交”结构为特征。因此,我们得出结论,已知由5催化的环加成和环异构化反应并非始于1,3-二烯基序,而是需要一个烯炔实体作为起始单元。


























 京公网安备 11010802027423号
京公网安备 11010802027423号