European Journal of Medicinal Chemistry ( IF 6.7 ) Pub Date : 2017-08-24 , DOI: 10.1016/j.ejmech.2017.08.046 Agnese C. Pippione , Alessandro Giraudo , Davide Bonanni , Irene M. Carnovale , Elisabetta Marini , Clara Cena , Annalisa Costale , Daniele Zonari , Klaus Pors , Maria Sadiq , Donatella Boschi , Simonetta Oliaro-Bosso , Marco L. Lolli
|
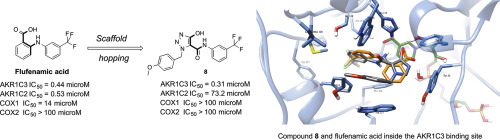
The aldo-keto reductase 1C3 isoform (AKR1C3) plays a vital role in the biosynthesis of androgens, making this enzyme an attractive target for castration-resistant prostate cancer therapy. Although AKR1C3 is a promising drug target, no AKR1C3-targeted agent has to date been approved for clinical use. Flufenamic acid, a non-steroidal anti-inflammatory drug, is known to potently inhibit AKR1C3 in a non-selective manner as COX off-target effects are also observed. To diminish off-target effects, we have applied a scaffold hopping strategy replacing the benzoic acid moiety of flufenamic acid with an acidic hydroxyazolecarbonylic scaffold. In particular, differently N-substituted hydroxylated triazoles were designed to simultaneously interact with both subpockets 1 and 2 in the active site of AKR1C3, larger for AKR1C3 than other AKR1Cs isoforms. Through computational design and iterative rounds of synthesis and biological evaluation, novel compounds are reported, sharing high selectivity (up to 230-fold) for AKR1C3 over 1C2 isoform and minimal COX1 and COX2 off-target inhibition. A docking study of compound 8, the most interesting compound of the series, suggested that its methoxybenzyl substitution has the ability to fit inside subpocket 2, being involved in π-π staking interaction with Trp227 (partial overlapping) and in a T-shape π-π staking with Trp86. This compound was also shown to diminish testosterone production in the AKR1C3-expressing 22RV1 prostate cancer cell line while synergistic effect was observed when 8 was administered in combination with abiraterone or enzalutamide.
中文翻译:
羟三唑衍生物作为有效的和选择性的醛酮还原酶1C3(AKR1C3)抑制剂,通过生物等排支架跳跃方法发现
醛酮还原酶1C3亚型(AKR1C3)在雄激素的生物合成中起着至关重要的作用,使该酶成为去势抵抗性前列腺癌治疗的诱人靶标。尽管AKR1C3是有前途的药物靶标,但迄今为止尚未批准任何AKR1C3靶向剂用于临床。氟苯那酸是一种非甾体类抗炎药,已知会以非选择性方式有效抑制AKR1C3,因为还观察到了COX脱靶作用。为了减少脱靶效应,我们应用了一种支架跳跃策略,将氟苯那酸的苯甲酸部分替换为酸性羟唑羰基支架。特别是N取代的羟基化三唑被设计为同时与AKR1C3活性位点中的亚位1和2相互作用,对于AKR1C3而言,它比其他AKR1Cs同工型更大。通过计算设计以及合成和生物学评估的迭代轮次,报道了新型化合物,它们对AKR1C3的选择性高于1C2亚型(高达230倍),并且对COX1和COX2的脱靶抑制作用最小。化合物8的对接研究,该系列中最有趣的化合物,表明其甲氧基苄基取代基能够嵌入子链2内,参与与Trp227的π-π赌注相互作用(部分重叠),并与Trp86呈T形π-π赌注。还显示了该化合物减少了表达AKR1C3的22RV1前列腺癌细胞系中的睾丸激素生成,而当将8与阿比特龙或恩杂鲁胺合用时,则观察到了协同作用。

























 京公网安备 11010802027423号
京公网安备 11010802027423号