当前位置:
X-MOL 学术
›
J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Exploring the Role of N6-Substituents in Potent Dual Acting 5′-C-Ethyltetrazolyladenosine Derivatives: Synthesis, Binding, Functional Assays, and Antinociceptive Effects in Mice∇
Journal of Medicinal Chemistry ( IF 7.3 ) Pub Date : 2017-04-27 00:00:00 , DOI: 10.1021/acs.jmedchem.7b00291 Riccardo Petrelli 1 , Mirko Scortichini 1 , Sonja Kachler 2 , Serena Boccella 3 , Carmen Cerchia 4 , Ilaria Torquati 1 , Fabio Del Bello 1 , Daniela Salvemini 5 , Ettore Novellino 4 , Livio Luongo 3 , Sabatino Maione 3 , Kenneth A. Jacobson 6 , Antonio Lavecchia 4 , Karl-Norbert Klotz 2 , Loredana Cappellacci 1
Journal of Medicinal Chemistry ( IF 7.3 ) Pub Date : 2017-04-27 00:00:00 , DOI: 10.1021/acs.jmedchem.7b00291 Riccardo Petrelli 1 , Mirko Scortichini 1 , Sonja Kachler 2 , Serena Boccella 3 , Carmen Cerchia 4 , Ilaria Torquati 1 , Fabio Del Bello 1 , Daniela Salvemini 5 , Ettore Novellino 4 , Livio Luongo 3 , Sabatino Maione 3 , Kenneth A. Jacobson 6 , Antonio Lavecchia 4 , Karl-Norbert Klotz 2 , Loredana Cappellacci 1
Affiliation
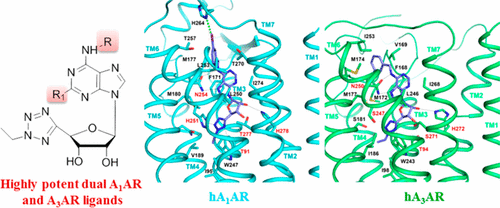
|
Structural determinants of affinity of N6-substituted-5′-C-(ethyltetrazol-2-yl)adenosine and 2-chloroadenosine derivatives at adenosine receptor (AR) subtypes were studied with binding and molecular modeling. Small N6-cycloalkyl and 3-halobenzyl groups furnished potent dual acting A1AR agonists and A3AR antagonists. 4 was the most potent dual acting human (h) A1AR agonist (Ki = 0.45 nM) and A3AR antagonist (Ki = 0.31 nM) and highly selective versus A2A; 11 and 26 were most potent at both h and rat (r) A3AR. All N6-substituted-5′-C-(ethyltetrazol-2-yl)adenosine derivatives proved to be antagonists at hA3AR but agonists at the rA3AR. Analgesia of 11, 22, and 26 was evaluated in the mouse formalin test (A3AR antagonist blocked and A3AR agonist strongly potentiated). N6-Methyl-5′-C-(ethyltetrazol-2-yl)adenosine (22) was most potent, inhibiting both phases, as observed combining A1AR and A3AR agonists. This study demonstrated for the first time the advantages of a single molecule activating two AR pathways both leading to benefit in this acute pain model.
中文翻译:

探索N 6-取代基在有效的双重作用5'- C-乙基四唑基腺苷衍生物中的作用:小鼠的合成,结合,功能测定和抗伤害感受作用∇
利用结合和分子模型研究了N 6-取代的5'- C-(乙基四唑-2-基)腺苷和2-氯腺苷衍生物在腺苷受体(AR)亚型上的亲和力的结构决定因素。小的N 6-环烷基和3-卤代苄基提供了有效的双作用A 1 AR激动剂和A 3 AR拮抗剂。4是最有效的双重作用人类(h)A 1 AR激动剂(K i = 0.45 nM)和A 3 AR拮抗剂(K i = 0.31 nM),相对于A 2A具有高度选择性;11和26在h和大鼠(r)A 3 AR上均最有效。所有的N 6-取代的5'- C-(乙基四唑-2-基)腺苷衍生物在hA 3 AR处均被证明是拮抗剂,而在rA 3 AR处则为激动剂。的镇痛11,22,和26在小鼠进行评价福尔马林试验(A 3 AR拮抗剂阻止和A 3 AR激动剂强烈强化)。如结合A 1 AR和A 3观察到的,N 6-甲基-5'- C-(乙基四唑-2-基)腺苷(22)最有效,抑制了两个相。AR激动剂。这项研究首次证明了单个分子激活两条AR通路的优势,都可在这种急性疼痛模型中受益。
更新日期:2017-05-06
中文翻译:
探索N 6-取代基在有效的双重作用5'- C-乙基四唑基腺苷衍生物中的作用:小鼠的合成,结合,功能测定和抗伤害感受作用∇
利用结合和分子模型研究了N 6-取代的5'- C-(乙基四唑-2-基)腺苷和2-氯腺苷衍生物在腺苷受体(AR)亚型上的亲和力的结构决定因素。小的N 6-环烷基和3-卤代苄基提供了有效的双作用A 1 AR激动剂和A 3 AR拮抗剂。4是最有效的双重作用人类(h)A 1 AR激动剂(K i = 0.45 nM)和A 3 AR拮抗剂(K i = 0.31 nM),相对于A 2A具有高度选择性;11和26在h和大鼠(r)A 3 AR上均最有效。所有的N 6-取代的5'- C-(乙基四唑-2-基)腺苷衍生物在hA 3 AR处均被证明是拮抗剂,而在rA 3 AR处则为激动剂。的镇痛11,22,和26在小鼠进行评价福尔马林试验(A 3 AR拮抗剂阻止和A 3 AR激动剂强烈强化)。如结合A 1 AR和A 3观察到的,N 6-甲基-5'- C-(乙基四唑-2-基)腺苷(22)最有效,抑制了两个相。AR激动剂。这项研究首次证明了单个分子激活两条AR通路的优势,都可在这种急性疼痛模型中受益。


























 京公网安备 11010802027423号
京公网安备 11010802027423号