当前位置:
X-MOL 学术
›
Chem. Rev.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations
Chemical Reviews ( IF 62.1 ) Pub Date : 2017-02-13 00:00:00 , DOI: 10.1021/acs.chemrev.6b00590 Abhik Ghosh 1
Chemical Reviews ( IF 62.1 ) Pub Date : 2017-02-13 00:00:00 , DOI: 10.1021/acs.chemrev.6b00590 Abhik Ghosh 1
Affiliation
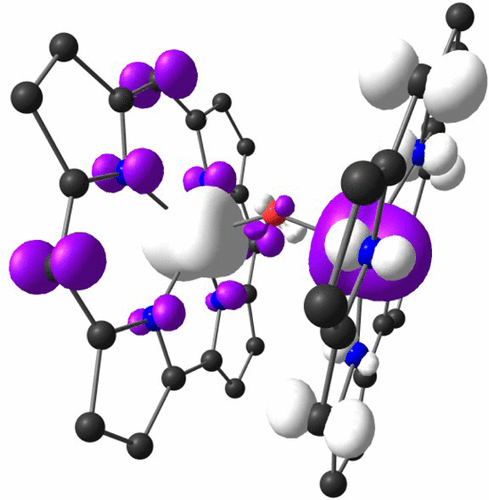
|
Presented herein is a comprehensive account of the electronic structure of corrole derivatives. Our knowledge in this area derives from a broad range of methods, including UV–vis–NIR absorption and MCD spectroscopies, single-crystal X-ray structure determination, vibrational spectroscopy, NMR and EPR spectroscopies, electrochemistry, X-ray absorption spectroscopy, and quantum chemical calculations, the latter including both density functional theory and ab initio multiconfigurational methods. The review is organized according to the Periodic Table, describing free-base and main-group element corrole derivatives, then transition-metal corroles, and finally f-block element corroles. Like porphyrins, corrole derivatives with a redox-inactive coordinated atom follow the Gouterman four-orbital model. A key difference from porphyrins is the much wider prevalence of noninnocent electronic structures as well as full-fledged corrole•2– radicals among corrole derivatives. The most common orbital pathways mediating ligand noninnocence in transition-metal corroles are the metal(dz2)–corrole(“a2u”) interaction (most commonly observed in Mn and Fe corroles) and the metal(dx2–y2)–corrole(a2u) interaction in coinage metal corroles. Less commonly encountered is the metal(dπ)–corrole(“a1u”) interaction, a unique feature of formal d5 metallocorroles. Corrole derivatives exhibit a rich array of optical properties, including substituent-sensitive Soret maxima indicative of ligand noninnocence, strong fluorescence in the case of lighter main-group element complexes, and room-temperature near-IR phosphorescence in the case of several 5d metal complexes. The review concludes with an attempt at identifying gaps in our current knowledge and potential future directions of electronic–structural research on corrole derivatives.
中文翻译:

Corrole衍生物的电子结构:分子结构,光谱学,电化学和量子化学计算的见解
本文介绍了对Corrole衍生物的电子结构的全面描述。我们在这方面的知识来自广泛的方法,包括UV-vis-NIR吸收和MCD光谱,单晶X射线结构测定,振动光谱,NMR和EPR光谱,电化学,X射线吸收光谱和量子化学计算,后者包括密度泛函理论和从头算多构型方法。审查是根据元素周期表组织的,描述了游离碱和主族元素的衍生物,然后是过渡金属的元素,最后是f嵌段元素的元素。像卟啉一样,带有氧化还原惰性配位原子的腐蚀衍生物也遵循Gouterman四轨道模型。•2 –腐蚀衍生物中的自由基。过渡金属分子中最常见的介导配体非纯净的轨道途径是金属(d z 2)-分子(“ a 2u ”)相互作用(最常见于锰和铁分子中)和金属(d x 2- y 2)。)-造币金属腐蚀中的corrole(a 2u)相互作用。少通常遇到的是金属(d π)-corrole(“一个1U ”)相互作用,正式d的一个独特的特征5金属茂。Corrole衍生物具有丰富的光学特性,包括对取代基敏感的Soret最大值,表明配体无毒,在较轻的主族元素配合物的情况下具有强荧光,在几种5d金属配合物的情况下具有室温近红外磷光。这篇综述的最后是试图找出我们目前在腐蚀衍生物的电子结构研究方面的知识和潜在的未来方向的差距。
更新日期:2017-02-13
中文翻译:
Corrole衍生物的电子结构:分子结构,光谱学,电化学和量子化学计算的见解
本文介绍了对Corrole衍生物的电子结构的全面描述。我们在这方面的知识来自广泛的方法,包括UV-vis-NIR吸收和MCD光谱,单晶X射线结构测定,振动光谱,NMR和EPR光谱,电化学,X射线吸收光谱和量子化学计算,后者包括密度泛函理论和从头算多构型方法。审查是根据元素周期表组织的,描述了游离碱和主族元素的衍生物,然后是过渡金属的元素,最后是f嵌段元素的元素。像卟啉一样,带有氧化还原惰性配位原子的腐蚀衍生物也遵循Gouterman四轨道模型。•2 –腐蚀衍生物中的自由基。过渡金属分子中最常见的介导配体非纯净的轨道途径是金属(d z 2)-分子(“ a 2u ”)相互作用(最常见于锰和铁分子中)和金属(d x 2- y 2)。)-造币金属腐蚀中的corrole(a 2u)相互作用。少通常遇到的是金属(d π)-corrole(“一个1U ”)相互作用,正式d的一个独特的特征5金属茂。Corrole衍生物具有丰富的光学特性,包括对取代基敏感的Soret最大值,表明配体无毒,在较轻的主族元素配合物的情况下具有强荧光,在几种5d金属配合物的情况下具有室温近红外磷光。这篇综述的最后是试图找出我们目前在腐蚀衍生物的电子结构研究方面的知识和潜在的未来方向的差距。


























 京公网安备 11010802027423号
京公网安备 11010802027423号